
Does evolution conserve software code in a similar way to genes?
Utility-functions in our software and biology

A common analogy of a DNA sequence is that it’s the “code” for a biological system. This code is under constant mutation pressure, an accumulated history of random mutation events change the sequence over time. When random mutations lead to deleterious outcomes, cells are less likely to survive and propagate the change into the future. Therefore, a section of DNA which remains unchanged or conserved for a long period of time is more likely to have an important function.
I was interested in whether or not this analogy would hold in a real software codebase. If I looked the accumulated history of changes, would the conserved parts be the most important? In a software codebase git allows us to see a history of changes (or mutations), and we can search for analogous functions which are preserved overtime. I wrote some code to do that here.
If we utilize this history to find the most “evolutionarily preserved code”, what does it look like in open source software? Turns out it looks like a utility function. ~60% of the top ranked “conserved” functions are utility functions which have a re-usable general purpose across the codebase, but are not directly aligned with the library’s goal.
What about in our cells? It’s basically the same! 56%-65% of the top 1000 most conserved genes are “housekeeping” or utility genes expressed across all cells, independent of function. In both the biological case and the software case these utility-esque functions/genes over-index relative to the baseline rate in the genome/codebases as a whole, so there is a real correlation between being conserved and being a utility function.
It’s unclear if this represents a broader complexity trend, but it’s pretty wild that the most conserved “code” in software and biology have an analogous purpose.
How to find evolutionary conserved code
(you can skip this section if you just want to see the results, they are understandable without it)
This biological analogy isn’t perfect, so I had to make some simplifying assumptions. Firstly, changes to a codebase aren’t really random, they are biased towards new features or bug-fixes, and usually recorded in coherent units (commits). I considered each individual commit to a codebase as a single mutation “trial”. Commits are tracked via each individual line changed, so, as a simplifying assumption, I only considered a “function” as the smallest unit that we would analyze. In each recorded commit, I kept a database of whether a function materially changed or not. (I tried to skip trivial things like renames or docstring updates)
We can make a pretty simple metric for evolutionary conservation of each recorded function, the ratio of un-altering commits to total commits, or trials. I would like to model the uncertainty of this ratio, because a function which has survived without changes in 100 commits is much more stable than one which has survived unchanged after 2 commits. So I modeled each trial for function f as a Bernoulli event with unknown survival probability p. Using a Beta conjugate prior over p as a function of k (unchanged outcomes) in n (total trials) gives us a posterior distribution like this:
where,
α, β are Beta prior parameters (default 1, 1; uniform)
n is the total trials/commits for a function f
k is the total trials/commits where f survived unchanged
To properly rank the posterior distribution, I used the lower credible bound (p=0.05), which captures uncertainty as a single number. So that brings us to our evolutionary quality metric (EQM).
Awesome — but, commits are often isolated to a single feature, so we can’t really consider each commit as contributing to a trial (n) for every single function f. I needed some logic to determine when a commit “counted” as an attempted mutation. My approach here was to use the AST parsed call graph of each function to determine if either the function itself was altered or if any caller function was altered. I only traversed a single node in this call graph. The logic is that if a developer modified or added a calling function, this would be the circumstance that would create the most mutation pressure.
Now with this metric, we can rank every function in large open source codebases to look at the most “conserved” functions. (see code here)
Conserved Code Examples
What does code look like when we examine the top ranked conserved functions? Let’s check out some popular open source projects.
Let’s start with FastAPI, where the top ranked most preserved function is a very simple utility function, get_path_param_names.
def get_path_param_names(path: str) -> set[str]:
return set(re.findall("{(.*?)}", path))This elegant little function has been subjected to 46 different commits where it needed to be used. And yet, it only needed to change 4 times. There are several other utility functions in the top 50 (19 of them in fact). Looking at the scikit-learn library, a top ranked function is assert_allclose. A “close” comparision assertion for floating point values which is dtype aware, so that the the relative tolerance parameter can scale with the input values.
def assert_allclose(
actual, desired, rtol=None, atol=0.0, equal_nan=True, err_msg="", verbose=True
):
"""dtype-aware variant of numpy.testing.assert_allclose
This variant introspects the least precise floating point dtype
in the input argument and automatically sets the relative tolerance
parameter to 1e-4 float32 and use 1e-7 otherwise (typically float64
in scikit-learn).
`atol` is always left to 0. by default. It should be adjusted manually
to an assertion-specific value in case there are null values expected
in `desired`.
The aggregate tolerance is `atol + rtol * abs(desired)`.
Parameters
----------
actual : array_like
Array obtained.
desired : array_like
Array desired.
rtol : float, optional, default=None
Relative tolerance.
If None, it is set based on the provided arrays' dtypes.
atol : float, optional, default=0.
Absolute tolerance.
equal_nan : bool, optional, default=True
If True, NaNs will compare equal.
err_msg : str, optional, default=''
The error message to be printed in case of failure.
verbose : bool, optional, default=True
If True, the conflicting values are appended to the error message.
Raises
------
AssertionError
If actual and desired are not equal up to specified precision.
See Also
--------
numpy.testing.assert_allclose
Examples
--------
>>> import numpy as np
>>> from sklearn.utils._testing import assert_allclose
>>> x = [1e-5, 1e-3, 1e-1]
>>> y = np.arccos(np.cos(x))
>>> assert_allclose(x, y, rtol=1e-5, atol=0)
>>> a = np.full(shape=10, fill_value=1e-5, dtype=np.float32)
>>> assert_allclose(a, 1e-5)
"""
dtypes = []
actual, desired = np.asanyarray(actual), np.asanyarray(desired)
dtypes = [actual.dtype, desired.dtype]
if rtol is None:
rtols = [1e-4 if dtype == np.float32 else 1e-7 for dtype in dtypes]
rtol = max(rtols)
np_assert_allclose(
actual,
desired,
rtol=rtol,
atol=atol,
equal_nan=equal_nan,
err_msg=err_msg,
verbose=verbose,
)This function has had modified or added callers 527 times, and it has only been modified 1 time! That means it’s not ever needed a material change since its been written, but it was useful to developers in over 500 other commits. Great ROI. So is this utility function theme generally true?
I used an LLM to categorize each function in the top-200 evolutionarily conserved ranking across 4 large open source Python libraries. Then I looked at the fraction considered “general purpose utilities used across the library”. Results below:
Utility functions in the top 50 and top 200 ranked by evolutionary conservation
FastAPI, top-50: 19/50 = 38%, top-200: 37/200 = 19%
Pydantic, top-50: 36/50 = 72%, top-200: 87/200 = 44%
Scikit-learn, top-50: 42/50 = 84%, top-200: 113/200 = 57%
SQLAlchemy, top-50: 23/50 = 46%, top-200: 54/200 = 27%
Total, top-50: 120/200 = 60%, top-200: 291/800 = 36%
So the density of utility functions is front-loaded amongst the most conserved functions in each codebase. In general these functions appear to be the “survivors” across 1000s of commits and 100s of contributors in these libraries.
So basically — utility functions dominate the landscape of conserved code. I think this makes sense. They are often pure functions to make things more easily composable. They are often written to improve the developer experience in service of a new feature, so there is a high likelihood of re-use. Finally, since they are usually outside of the path of a libraries “application logic”, they are less likely to change as scope creed happens.
You can examine your own open source repos with my analysis codebase here.
So… does this same trend hold in biology?
Biological Comparison
So utility functions are our software survivors — this begs the question as to whether conserved genes will create proteins that are also utility functions. We can look for analogous utility genes that serve general-purpose, always-on cellular roles rather than specialized tissue-specific ones.
Just like in our software example, we need to rank genes by how strongly evolution has resisted changes to their protein sequence. Basically, we look at two reference genomes, one for humans and one for mice. Since humans and mice have diverged evolutionarily (~90 million years ago), we can search for “orthologs”, genes which came from the same ancestral gene, and compare them. Each nucleotide site in the sequence will be relatively synonymous (i.e. changes to the site don’t change the codon/protein) or nonsynonymous (i.e. changes to the site will change the codon/protein). Since mutations happen randomly, site differences between mouse and human genomes in synonymous sites help us establish a baseline rate of mutation within each gene. Substitutions between mouse and human genomes in nonsynonymous sites show us where the genes materially “changed”. If you add up all the sites in a gene, and divide by the ratio of nonsynonymous substitutions per nonsynonymous site to the ratio of synonymous substitutions per synonymous site (a metric called dN/dS), you can quantify how evolutionarily “conserved” a gene is.
If dN is much lower than dS (dN/dS << 1), that means mutations that would change the protein are being removed by natural selection. Lower dN/dS means stronger conservation. We used dN/dS values from Ensembl Compara Release 95, restricted to one-to-one human-mouse orthologs. This gave us about 15,000 genes with valid dN/dS values. So, just like in our software slices, I ranked every gene and took the top 1,000 conserved genes to look at what fraction would be utility-esque.
I based my utility categorization on the Human Protein Atlas (HPA) RNA expression data across human tissues. I considered two thresholds, strict housekeeping (“low tissue specificity”) meaning it is expressed at similar levels across all tissues. These are the closest analogy to a utility function in software. Examples include ribosomal proteins, metabolic enzymes like GAPDH, and cytoskeletal components.
My second threshold was for broadly expressed genes (“detected in all”), so levels may vary in tissues, but they are always present. This is more permissive, but these genes likely serve some kind of utility purpose, used more heavily in some tissues than others. See my plot below of expression rates of these categories (from ensembl) ranked by conservation ordering.
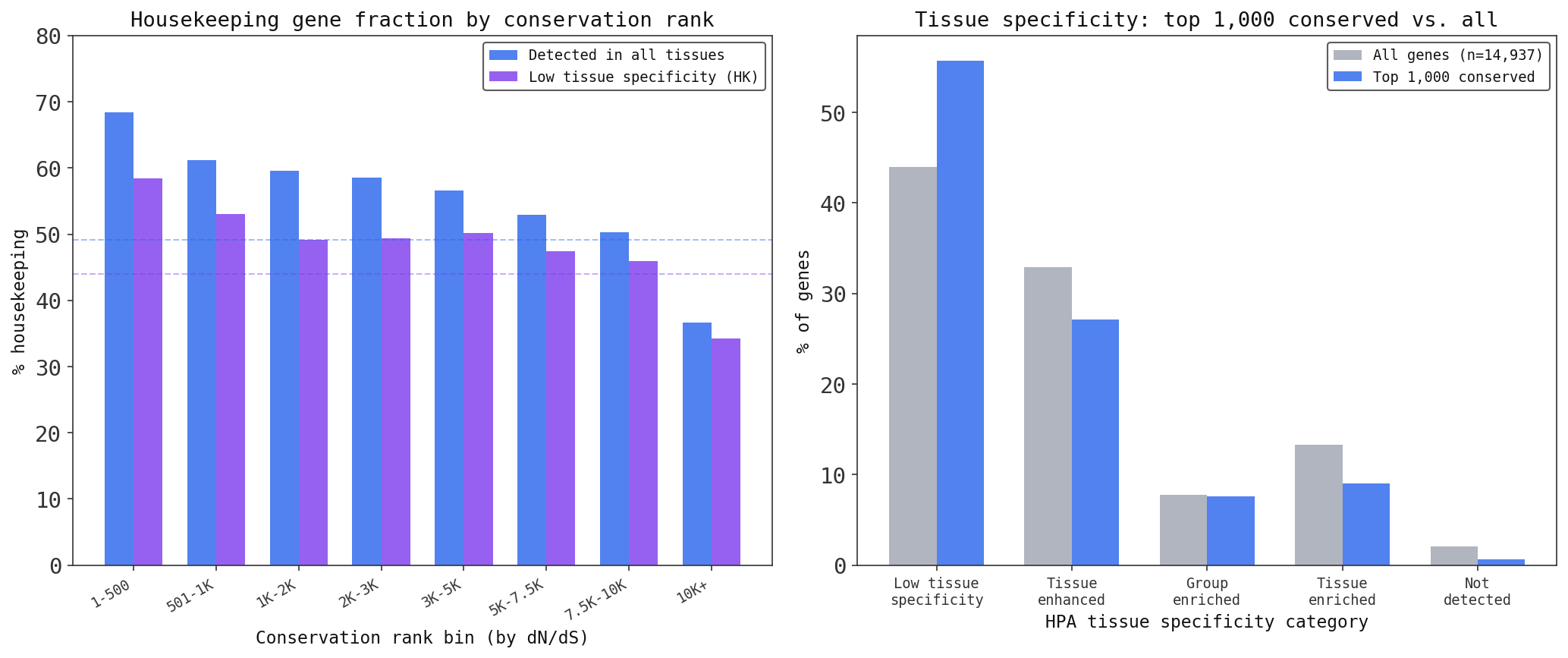
To level set, the genome-wide baseline rates are ~44% for my strict housekeeping definition and ~49% broadly expressed.
Finally! Among the top 1,000 most conserved genes, 56% are in my strict housekeeping category and 65% are broadly expressed. This over-indexes relative to the genome-wide baselines of 44% and 49%, confirming that highly conserved genes are more likely to be general-purpose utilities. Just like in our software samples!
What does it all mean?
Maybe this is obvious. An object that interacts with multiple components of a system should be more important than an object that interacts with a single source. But that being said, an object that is utilized in many different places should also be under a lot of pressure to change as well. Maybe at some basal level, utility functions are able to capture invariants in “function-space” more easily. Maybe when you write utility functions, it’s simpler to capture all the requirements. I’m not really sure.
I just think it’s interesting that the “purpose” of the top conserved genes and software ends up looking the same.